Coping with stress is essential for all organisms, and several stress responses have evolved to maintain homeostasis. Senescence and autophagy are two of the most critical stress responses as (1) they respond to various types of stress and (2) they are closely associated with numerous age-related diseases and aging. Our overarching research goals are (1) to systematically identify the components that make up their regulatory networks; (2) to mechanistically understand the cellular and molecular logic of these systems; and (3) to define the nature of their dysregulation in age-related diseases and aging. In other words, We wish to understand how senescence and autophagy operate, why they are built the way they are, and what the consequence of their dysregulation is. We employ genetics, molecular biology, biochemistry, genomics, and cell biology with new technologies developed in our lab [e.g., SIP (selective autophagy substrates identification platform), LysoIP-DEG (immunopurified lysosomes followed by density gradient ultracentrifugation)] to advance these goals. A better understanding of these complex stress responses will provide new insights into the fundamental question of why we age. In principle, it will help the development of new therapeutic strategies for aging and age-related diseases, enhancing human health.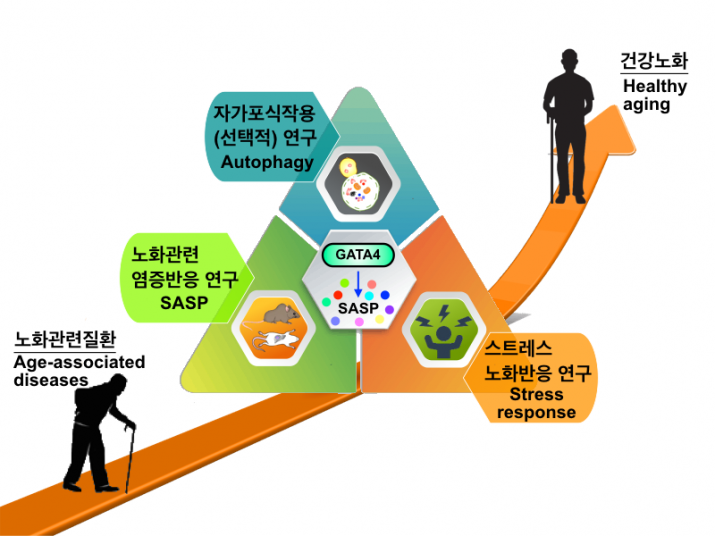
Cellular senescence is a terminal program of growth arrest triggered by many stresses. Like apoptosis, it acts intrinsically as a tumor-suppressive mechanism by limiting further proliferation of damaged cells. Furthermore, in contrast to apoptosis that mainly acts cell-autonomously, cellular senescence can also function cell-nonautonomously to coordinate homeostasis responses; This occurs through the secretion of pro-inflammatory cytokines, growth factors, and proteases from senescent cells: the Senescence Associated Secretory Phenotype (SASP).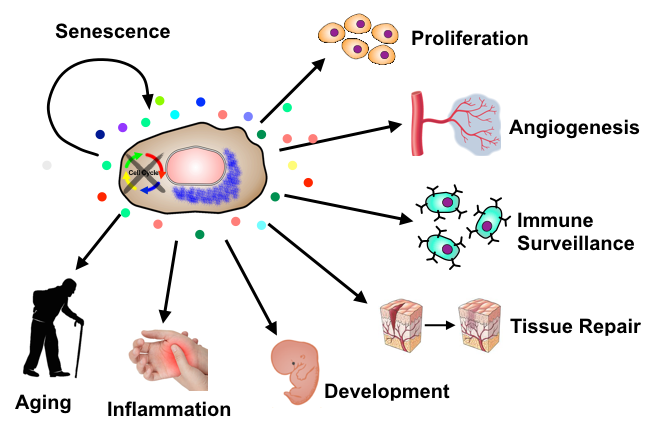
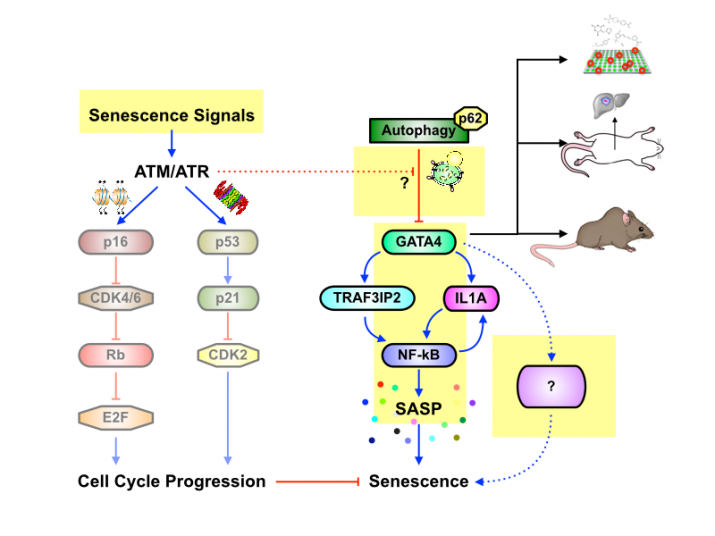
Despite such pleiotropic effects of the SASP, little is known about how it is regulated until recently, in contrast to well-studied senescence growth arrest where the p53 and p16 tumor suppressor pathways have a crucial role. Our recent study has identified a transcription factor GATA4 as a key switch to activate the SASP, independently from the p53 and p16 pathways. In addition, we have discovered how GATA4 is regulated during cellular senescence by selective autophagy and the DNA damage response (DDR). Thus, our study establishes the GATA4 pathway as a new branch in the senescence regulatory network and opens a new field that connects DDR, autophagy, protein stability, senescence, and inflammation.
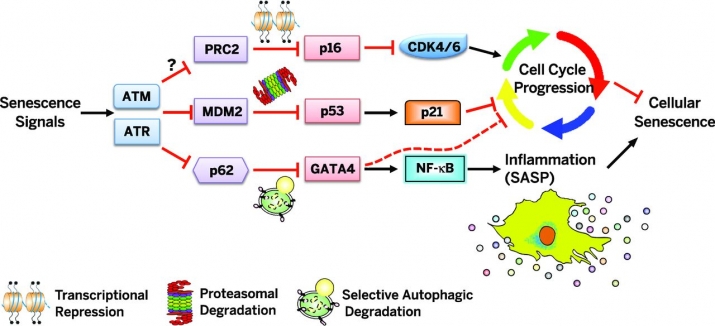
Although we gained new insight into the role of selective autophagy during senescence and SASP regulation through GATA4, our current understanding of the GATA4 pathway is still limited. We are currently employing genetics, molecular biology, biochemistry, genomics, and cell biology to elucidate 1) the selective autophagy network and 2) the senescence regulatory network with a focus on the SASP in detail. Specifically, we will 1) elucidate the GATA4 pathway in the senescence regulatory network, 2) determine potential in vivo roles of the GATA4-SASP pathway during tumorigenesis and aging, 3) examine the stress-specific regulation of selective autophagy, cellular senescence, and the SASP, and 4) develop the stress-specific reporter system that is suitable for genome-wide genetic screens.